بررسی ژنتیکی ناقلان و بیماران مبتلا به سیستیک فیبروزیس
آزمایش بررسی ژنتیکی ناقلان و بیماران مبتلا به سیستیک فیبروزیس :
◀︎این آزمایش در بخش مولکولی آزمایشگاه فجر انجام میپذیرد.
سیستیک فیبروزیس CF) Cystic Fibrosis) یک بیماری اتوزومی مغلوب است. این بیماری در نتیجه اختلال در انتقال یون های کلر درسلول های اپی تلیال به دلیل جهش هایی در ژن تنظیم گر هدایت غشایی (CF) (CFTR) ایجاد می شود. اندام هایی که بیشتر از بقیه در این بیماری درگیر می شوند، ریه و پانکراس می باشند.
بیماری ریوی مزمن همراه با عفونتهای مکرر در نهایت باعث تغییرات فیبروتیک در ریه همراه با نارسایی قلبی ثانویه می شود.
در ۸۵٪ افراد مبتلا، عملکرد پانکراس دچار آسیب می شود، که در اثر انسداد مجاری پانکراس توسط ترشحات غلیظ، ترشح آنزیم ها کاهش می یابد. این نقص با نارسائی در جذب غذا و ایجاد مدفوع چرب همراه است. بیش از ۹۵% مردان مبتلا به CF به دلیل بسته بودن مادرزادی مجرای وازدفران آزواسپرمی دارند و نابارورند.
بیماری عمدتا در سفیدپوستان اروپایی مشاهده می شود. والدین یک فرد بیمار، ناقل بیماری هستند و در کل ریسک تولد فرزند بیمار در هر بارداری برای آنها ۲۵% می باشد. البته باید به این نکته توجه نمود که تنوع جغرافیایی و نژادی درتوزیع جهش ها وجود دارد لذا در محاسبه ریسک تجربی باید به این موضوع توجه نمود.
ژنتیک بیماری سیستیک فیبروزیس
ژن CFTR بر روی کروموزوم ۷ در موقعیت q31-327 قرار دارد. این ژن ناحیه ای از ژنوم با اندازه ای حدود kb250 را دربرگرفته و دارای ۲۷ اگزون می باشد. پروتئین حاصل از این ژن دارای ۱۴۸۰ اسید آمینه است.
تقریبا ۲۰۰۰ جهش در ژن CFTR تعیین شده است که شایع ترین آن حذف سه نوکلئوتیدی مجاور هم در موقعیت کدون ۵۰۸ می باشد که باعث حذف اسید آمینه فنیل آلانین از پروتئین می شود.
تشخیص بیماری سیستیک فیبروزیس
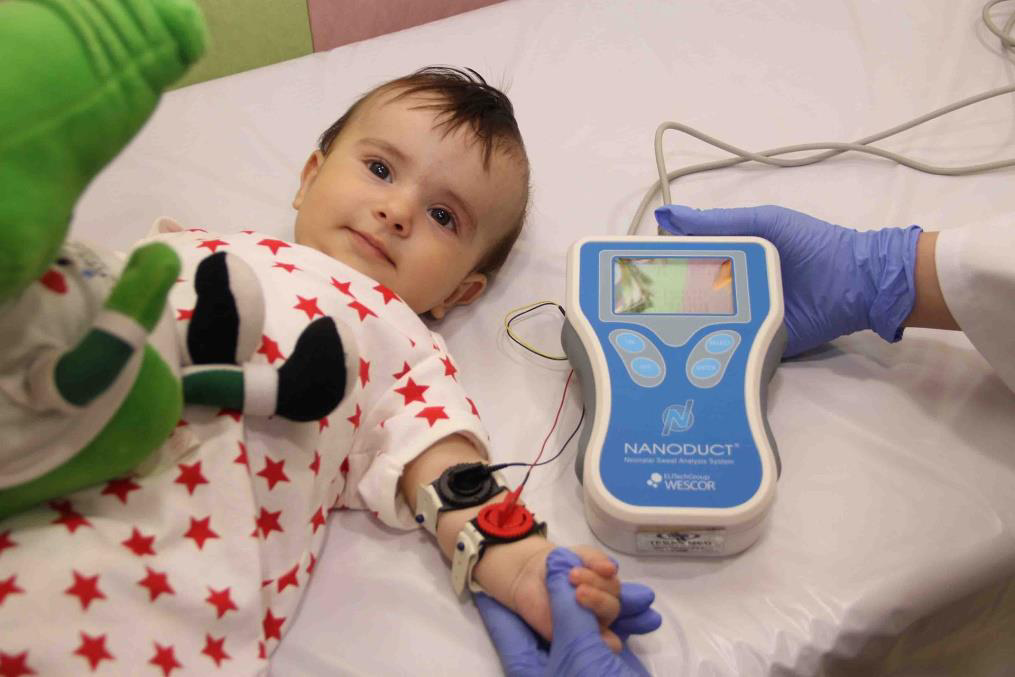
تست عرق : بررسی وجود سدیم کلرید درعرق ( >60 meq/IU)
آنالیز موتاسیون های ژنی: با توجه به نوع جهش می توان با روش های متفاوتی ژن CFTR را بررسی نمود.